逐次重合

逐次重合(ちくじじゅうごう、英: sequential polymerization)あるいは段階成長重合(だんかいせいちょうじゅうごう、step-growth polymerization) とは、官能基を二つ以上もつモノマーがまずダイマーを形成し、次にトライマー、と徐々に長いオリゴマーへと成長していき、最終的に長鎖ポリマーとなるような重合反応機構をいう。自然界に存在する多くのポリマーが、およびポリエステルやポリアミド、ポリウレタンなどのいくつかの合成ポリマーが逐次重合により生産されている。重合反応機構の性質から、大きな分子量に到達するまでには反応が高度に進行する必要がある。逐次重合の反応機構を思い描く最も簡単な方法は、複数の人間が手を繋ぎ合って人間の鎖を作る場面を想像することである。各人の二つの手が反応サイトに相当する。モノマーに二つよりも多い反応サイトが存在する場合もあり、この場合は分枝ポリマーが生成する。
歴史
人間に原始時代から用いられてきた自然ポリマーはほとんどが縮合型であった。初めての真の合成ポリマー材料はベークライトであり、1907年にレオ・ベークランドにより発表された。この材料はフェノールとホルムアルデヒドの逐次重合により生産される。1930年代デュポンにおける研究リーダーとして働いていた合成ポリマー科学の先駆者、ウォーレス・カロザースは逐次重合を用いる新たなポリエステルの製法を開発した。この反応は大きな分子量を達成することを目的として設計された初めての反応であり、かつ科学的理論によって結果を予言した上で行なわれた初めての重合反応でもある。カロザースは逐次重合系のふるまいを記述する一連の数式を開発し、今日ではカロザース方程式として知られている。物理化学者のポール・フローリーとの協力により、速度論や量論、分子量分布などの逐次重合のより詳しい数学的側面を記述する理論が開発された。カロザースはナイロンの発明者としても知られている。
逐次重合と縮合重合
逐次重合と縮合重合とは別の概念であり、必ずしも一致する概念ではない。実際、ポリウレタン重合時の反応機構は(他の低分子を生成しないので)付加重合であると言えるが、同時に逐次重合の反応機構により進行する。
付加重合と縮合重合の区別は1929年にカロザースにより次のように重合の生成物に基く区分として導入された[1][2]。
- ポリマーのみ:付加重合
- ポリマーと低分子:縮合重合
逐次重合と連鎖重合の区別は1953年にフローリーにより次のように反応機構に基く区分として導入された[3]。
逐次重合と連鎖重合との違い
逐次重合の特徴を示すため、しばしば連鎖重合との対比が行なわる。
| 逐次重合 | 連鎖重合 |
|---|---|
| 基質全体で成長が進行 | 各分子鎖の一端もしくは両端でのみ成長が進行 |
| モノマーは反応の初期段階で消滅 | 反応進行中の長いあいだモノマーが残存 |
| 類似の段階的成長が反応過程全体にわたって繰り返される | 開始反応、伝播反応、停止反応、連鎖移動の異る段階を踏んで進行 |
| 低転換率では平均分子量はゆっくりと増大し、長い分子鎖長を得るためには高度に反応が進行する必要がある | 主鎖分子量は反応中の速い段階で迅速に増加し、重合反応中にわたっておおよそ変化しない |
| 活性を保ったまま終了(停止しない) | 停止反応後には活性がなくなる |
| 重合開始剤の必要がない | 重合開始剤を必要とする |
様々な逐次重合による生成物
- ポリエステルは高いガラス転移温度 Tg と高い融点 Tm を持ち、およそ 175 °C までは良い機械物性を示し、溶媒や化学物質に対しての抵抗性が良い。繊維やフィルムの形で用いられる。繊維は織物、フェルト、タイヤコードなどに用いられ、フィルムは磁気記録媒体や高品質薄膜に用いられる。
- ポリアミド(ナイロン)は高い強度、弾性と耐摩擦性、耐久性、耐溶媒性などのバランスの良い物性を持つ。ポリアミドはロープ、ベルト、ファイバークロス、糸、ベアリングにおける金属代替材料、電線の被覆材などに応用される。
- ポリウレタンは耐摩擦性、硬さ、潤滑油への耐性、弾性を備えたエラストマーとしてや、反発性の優れた繊維として、溶媒や摩擦に対する耐久性を備えた被覆材として、強度と反発性、耐衝撃性を備えた発泡材として用いられる。
- ポリウレア(英語版)は高い Tg、油脂および溶媒への耐久性を示す。荷台のライニング材や橋梁塗装材、コーキング材や装飾デザインに用いられる。
- ポリシロキサンは液状、脂状、蝋状、樹脂状、ゴム状と幅広い物理的状態で利用しうる。用途としては消泡剤や剥離剤、ガスケット、封止材、ケーブルやワイヤーの絶縁材、高温液体・液体の導管などが挙げられる。
- ポリカーボネートは透明な自己消火性のある材料である。高い衝撃強度、熱・酸化に対する安定性など、熱可塑性結晶に類似した物性を有する。機械や自動車産業、 医療用などに用いられる。例えば、F-22戦闘機は高光学品質ポリカーボネートをコックピットキャノピー(英語版)に採用している。
- ポリスルフィドは顕著な耐油・耐溶媒性、気体不透過性、エージング耐性、オゾン耐性を有する。しかし、悪臭を持ち、引っ張り強度が低く、熱・酸化に対する耐久性も低い。ガソリンホースやガスケット、溶媒および気体への耐性が要求される部位に使用できる。
- ポリエーテルは熱可塑性、水溶性、様々な優良機械特性、あるていどの強度と剛性を示す。木綿や合成繊維の糊付け、粘着剤の安定化剤、結着剤、医薬品の成膜などに用いられる。
- ベークライトは耐熱性、形状安定性、ほとんどの溶媒に対する耐久性を有する。また、誘電物性も優れている。この材料は典型的にはその誘電物性から電気製品、ラジオ、テレビ、自動車向けの塑造部品に用いられる。他にも、含浸紙、ワニス、壁材の装飾用ラミネートなどにも用いられる。
- ポリトリアゾールポリマーはアルキン基とアジド基を有するモノマーから製造される。モノマー単位はそれぞれ、1,3-双極子環化付加反応(英語版)、別名アジド-アルキンヒュスゲン環化付加反応(英語版)により生じる1,2,3-トリアゾール基により繋がれる。これらのポリマーは強度の高い樹脂状[6]もしくはゲル状となる[7]。末端アルキン基と末端アジド基を持つオリゴペプチドモノマーからは、エンドペプチダーゼがオリゴペプチド単位に作用することにより生分解性を持ったクリックペプチドポリマー(英語版)が作られる[8]。
分枝ポリマー
3つ以上の官能基を持つモノマーはポリマー中に分枝を生じさせ、究極的には架橋大規模構造、転換率が低くてもネットワーク構造を生じる。樹状トポロジーがネットワークトポロジーへと遷移する点は、突然の粘度変化を伴うのでゲル化点(英語版)と呼ばれる。初のいわゆる熱硬化性樹脂はベークライトである。逐次重合の過程ではいつも水が放出されるとは限らない。非環式ジエンメタセシス(英語版) (ADMET) ではジエンがエチレンの脱離を伴って重合する。
反応速度論
逐次重合における反応速度論をポリエステル化機構を例に説明する。単純なエステル化は酸のプロトン化に続いてアルコールが相互作用することによりエステルと水が生じる酸触媒過程である。しかし、この速度論モデルにはいくつかの仮定が必要である。最初の仮定は、水(もしくは他の脱離基)が効率的に除去されるというものである。次に、官能基の反応性は鎖長に依存しないと仮定する。最後に、各ステップには1つのアルコールと1つの酸のみが関与すると仮定する。
![{\displaystyle {\frac {1}{1-p^{n-1}}}=1+(n-1)kt[{\ce {COOH}}]^{n-1}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f40d7c3649f67e96e10edcd26e6fab23275516ae)
これは重合度に対する一般的な速度法則である。ここで、n は反応次数を表わす。
自己触媒ポリエステル化
酸触媒が添加されない場合でも、反応は酸の自己触媒作用により進行する。したがってこの場合、任意の時刻 t における縮合速度は -COOH 基の消失速度から得られる。
![{\displaystyle rate={\frac {-d[{\ce {COOH}}]}{dt}}=k[{\ce {COOH}}]^{2}[{\ce {OH}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e36e9f9e72374e37c029a720efe9cb5e244de56e)
二次の [\ce{COOH}] 項は触媒作用に起因し、k は速度定数である。酸とグリコールが等量含まれる系では、官能基の濃度は次のようにシンプルに書ける。
![{\displaystyle rate={\frac {-d[{\ce {COOH}}]}{dt}}=k[{\ce {COOH}}]^{3}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c02467b42290958b886e503d42cab7b3882e6b9d)
カロザース方程式(英語版)を代入し積分すると、最終的に以下の形式を得る。
![{\displaystyle {\frac {1}{(1-p)^{2}}}=2kt[{\ce {COOH}}]^{2}+1=X_{n}^{2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/11bd2e3f368961bfa8f4002c38928e4755ed2cba)
自己触媒系では、数平均重合度 Xn は に比例して増加する[9]。

外部触媒ポリエステル化
触媒不在の反応は比較的遅く、高い Xn を得ることは容易ではない。触媒の存在下では反応速度が加速され、速度式は次のように書き換えられる[10]。
![{\displaystyle {\frac {-d[{\ce {COOH}}]}{dt}}=k[{\ce {COOH}}][{\ce {OH}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2fa3c69f4aa3a4c9646018d505e9e189a22e56ab)
このように、この反応は両方の官能基に対して一次反応である。したがって、
![{\displaystyle {\frac {-d[{\ce {COOH}}]}{dt}}=k[{\ce {COOH}}]^{2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/40623a5a6ecd32d96f118b41e932c376ed8c47f2)
積分すると次のようになる。
![{\displaystyle {\frac {1}{1-p}}=1+[{\ce {COOH}}]kt=X_{n}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8addf76f8424ddca5c944ab775c0d7186bf58703)
外部触媒系では、数平均重合度は に比例して増加する。

線形重合における分子量分布
重合生成物は分子量の異なる巨大分子の混合物である。 理論、実用の両面の理由から分子量の分布についての議論は興味を惹いている。分子量分布(英語版)はフローリーにより、官能基の反応性が等しいという考え方に基いた統計学的アプローチにより導からえた[11][12]。
確率
逐次重合はランダム過程であり、統計学を用いて x 個の構造単位を持つ分子鎖(x 量体)をみつける確率時間または転換率の関数としてを計算することができる。


官能基Aが既に反応している確率は

未反応のAを見付ける確率は

二つの式を組み合わせると

ここで、Px は x 量体が未反応のAを見付ける確率である。x が増えるにつれて確率は低下する。
数分率分布

数分率分布は系内における x 量体の比率であり、溶液中で x 量体を見つける確率と等しい。

N は反応中の存在するポリマー分子の総数である[13]。
重量分率分布

重量分率分布は、重量分率を用いて系内の x 量体の比率を表わしたものであり、質量で重み付けした発見確率である。

ここで、以下のような変数を用いた。
- Mo は繰り返し単位(英語版)のモル質量
- No はモノマー分子の初期総数
- N は未反応の官能基の個数
カロザース方程式(英語版)を代入すると、以下を得る。

さらに次も得られる。

多分散指数
多分散指数(英語版) (polydispersity index, PDI) は所与のポリマー試料の分子量分布の尺度の一つである。

しかし、逐次重合においてはカロザース方程式を用いてこの定義式を以下のように変形することができる。

したがって、逐次重合の場合は p=1 ならば PDI=2 と言える。
線形重合における分子量制御
量論制御の必要性
重合における分子量制御には二つの重要な面がある。ポリマーの物性は通常大きく分子量に依存するので、ポリマー合成においては特定の分子量の生成物を得る動機がある。所望の分子量よりも高くても低くても望ましくない。重合度は反応時間の関数であるから、適切な時期に反応をクエンチすることにより所望の分子量を得ることができる。しかし、こうして得られたポリマーには反応性を持っている官能基が残存するため、分子量が変化することがあり不安定である。
この状況は、二つのモノマーの濃度を若干量論比からずらし、反応物のうち片方を過剰に存在させることにより避けることができる。すると重合が片方のモノマーを完全に使い果すところまで進行したとき、全ての高分子鎖末端は過剰な側の官能基を持つことになる。これ以上重合が進むことは不可能となり、したがって後に分子量の変化は起こらず安定である。
所望の分子量を達成する別の方法として、官能基を一つだけ持つモノマーを少量添加する方法が挙げられる。単一官能基モノマーはしばしば連鎖停止剤とも呼ばれ、このモノマーが末端にあると官能基がなくなり、それ以上に重合することができないので、複官能基モノマーの重合を制御・制限することができる。
定量的側面
ポリマー分子量を適切に制御するには、複官能基モノマーの量論比からのずれもしくは単一官能基モノマーの比率を精密に調整する必要がある。量論比からのずれが大きすぎると分子量は小さすぎるものとなってしまう。したがって、反応物の量論比からのずれが分子量に及ぼす影響を定量的に知ることが重要となる。また、反応物の混合物中に最初から存在する、もしくは望ましくない副反応から生じるかもしれない不純物の効果も定量的に知る必要がある。反応に関与する官能基を持つ不純物が存在すると、それを定量的に考慮しないかぎり分子量が劇的に低くなってしまう。
より有用なことに、混合物中の反応物の量論比からのずれを精密に制御することで望ましい結果を得ることもできる。たとえば、ジアミンが酸クロリドに対して過剰な場合、やがて酸クロリドが完全に消費されると二つのアミン末端を持ち、それ以上反応しないポリアミドが得られる。このことはカロザース方程式を拡張することにより以下のように表現される。

ここで r は反応物分子の数比率である。
- ただし、NBB が過剰な側の分子数とする。

上式は単一官能基添加物の場合には次のように使うことができる。

ここで、NB は単一官能基添加物の数である。NB にかかる係数2は一つの分子Bは一つの過剰な分子B-Bと定量的に同じ効果を持つためである[14]。
多鎖重合
官能性が3のモノマーは重合に関与しうる3つの官能基を持つ。これによりポリマーは分枝を持つことになり、究極的には架橋大規模構造を形成する。3次元ネットワークが生じる点はゲル化点(英語版)として知られ、粘度の急激な変化により知ることができる。
より一般に、官能性指数 fav をモノマー単位ごとの平均官能基数として定義される。重合開始時点で N0 個の分枝を含み、官能基AとBが同数存在する系では、官能基の総数は N0fav となる。

そして、次の修正カロザース方程式を得る[15]。
- ここで p は


逐次重合ポリマーの進歩
既存の構成材料、特に金属を軽量で耐熱性のあるポリマーで置き換えることを目的として、新たなポリマーが設計されている。軽量ポリマーの利点は高い強度、対溶媒および化学的耐久性などが挙げられ、電気製品や自動車・航空機用エンジン部品、調理器具のコーティング、電子装置およびマイクロエレクトロニクスデバイスのコーティングや基板など、多様な潜在用途に寄与している。芳香族環ベースの高分子鎖は結合強度が高く、高分子鎖の剛性が高いため望ましい。高い分子量と架橋も同じ理由から望ましい。強い双極子-双極子相互作用、水素結合、結晶性(英語版)によっても耐熱性は向上する。所望の機械強度を得るには分子量が大きいことが要求されるが、溶解度が下がるという問題も引き起こす。この問題を解決するアプローチの一つとして、適切なモノマーおよびコモノマーを用いてイソプロピリデン基やC=O、SO2 を剛直ポリマーに導入して繋ぎ目を柔軟化する方法が挙げられる。他にも、末端に相互反応性官能基を持つ反応性テレケリックオリゴマーを合成し、オリゴマーを重合させることにより高い分子量を得るアプローチもあり、鎖延長と呼ばれる[16]。
芳香族ポリエーテル

様々な2, 6-二置換フェノールの、第一銅錯体塩触媒およびアミンを利用する酸化カップリングにより芳香族ポリエーテルが得られる。これは商業的にはポリ-p-フェニレンオキシド(英語版) (PPO) と呼ばれる。純粋なPPOは溶融体の粘度が高くあまり商業的に用いられない。PPOと耐衝撃性ポリスチレン (HIPS) の混合物が製品として入手可能である。
ポリエーテルスルホン

ポリエーテルスルホン (PES)はポリエーテルケトン、ポリスルホンとも呼ばれる。芳香族2ハロゲン化物とビスフェノラート塩との間の求核芳香族置換反応により合成される。ポリエーテルスルホンは部分的に結晶性を持ち、様々な水系・有期系環境で耐久性が高い。240-280 °C の温度範囲での継続利用に耐えられる。ポリケトンは自動車・航空宇宙産業、電気・電子ケーブル絶縁体などに用途がある。
芳香族ポリスルフィド
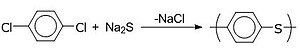
ポリ(p-フェニレンスルフィド) (PPS) は1-メチル-2-ピロリデン (NMP) などの極性溶媒中において硫化ナトリウムと p-ジクロロベンゼンを反応させて合成される。本質的に耐火性があり、有機系・水系環境で安定であるが、酸化剤に対して若干弱い。PPSは自動車や電子レンジ部品、フッ素樹脂と混合して調理器具のコーティング、バルブやパイプや電池の保護コーティングなどに用途がある[17]。
芳香族ポリイミド

芳香族ポリイミドはピロメチル酸無水物とp-フェニレンジアミンなどの二無水物とジアミンを反応させて合成される。ジアミンの代わりにジイソシアネートを使ってもよい。溶解度を考慮すると二無水物自体の代わりにその半酸半エステルの利用が勧められる場合もある。重合はポリイミドの不溶性のため、2段階にわけて行なわれる。1段めはNMPやN,N-ジメチルアセトアミドなどの極性非プロトン性溶媒中で可溶性・可融性の高分子量ポリアミック酸を生成する。できたポリアミック酸を処理して所望の物理的形状(フィルム、遷移、ラミネート、コーティング)の不溶・不融性の最終ポリマーを得る。
テレケリックオリゴマー法
テレケリック(英語版)オリゴマー法は、通常の重合様式を用いるが一般的に 50-3000 分子量のオリゴマー段階で反応を停止させる。単一官能性反応物は重合を制限するだけでなく、オリゴマーの末端に官能基を導入することができるので、これを後でオリゴマーのキュアリングのための反応に用いることができる。アルキン、ノルボルネン、マレイミド、ニトリト、シアネートなどの官能基がこの目的に利用される。マレイミドとノルボルネンにより末端キャップされたオリゴマーは熱によるキュアリングができる。アルキン、ニトリル、シアネートにより末端キャップされたオリゴマーは環化三量化(英語版)により芳香族構造を形成することができる。
関連項目
外部リンク
- Crosslinking
- The Chemical Heritage Foundation
出典
- ^ W. H. Carothers (1929). “STUDIES ON POLYMERIZATION AND RING FORMATION. I. AN INTRODUCTION TO THE GENERAL THEORY OF CONDENSATION POLYMERS”. Journal of the American Chemical Society 51 (8): 2548–2559. doi:10.1021/ja01383a041.
- ^ Paul J. Flory, "Principles of Polymer Chemistry", Cornell University Press, 1953, p.39.
- ^ Susan E. M. Selke, John D. Culter, Ruben J. Hernandez, "Plastics packaging: Properties, processing, applications, and regulations", Hanser, 2004, p.29.
- ^ Seymour, Raymond (1992). Polymer Chemistry An Introduction. Marcel Dekker, Inc.. ISBN 0-8247-8719-6
- ^ H.F. Mark, N. M. B., C. G. Overberger, G. Menges, Encyclopedia of Polymer Science and Engineering Wiley-Interscience: New York, 1988.
- ^ Wan, L.; Luo, Y.; Xue, L.; Tian, J.; Hu, Y.; Qi, H.; Shen, X.; Huang, F. et al. (2007). “Preparation and properties of a novel polytriazole resin. J. Appl. Polym”. Sci 104: 1038–1042. doi:10.1002/app.24849.
- ^ Yujing, L.; Liqiang, W.; Hao, Z; Farong, H.; Lei, D. (2013). “A novel polytriazole-based organogel formed by the effects of copper ions”. Polym. Chem. 4: 3444–3447. doi:10.1039/C3PY00227F.
- ^ Van Dijk, M.; Nollet, M.; Weijers, P.; Dechesne, A.; van Nostrum, C.; Hennink, W.; Rijkers, D.; Liskamp, R. (2008). “Synthesis and Characterization of Biodegradable Peptide-Based Polymers Prepared by Microwave-Assisted Click Chemistry”. Biomacromolecules 10: 2834–2843. doi:10.1021/bm8005984.
- ^ Yves Gnanou; Fontanille, M., Organic and Physical Chemistry of Polymers.
- ^ Cowie, J.M.G; Arrighi, V., Polymers: Chemistry and Physics of Modern Materials. 3 ed.; CRC Press: 2008.
- ^ Flory, Paul. Principles of Polymer Chemistry. Cornell University Press. pp. 321–322
- ^ Odian, George (1991). Principles of polymerization. John Wiley& Sons, INC. ISBN 0-471-61020-8
- ^ Stockmayer, Waters (1952). “Molecular distribution in condensation polymers”. Journal of polymer science IX (1): 69–71. Bibcode: 1952JPoSc...9...69S. doi:10.1002/pol.1952.120090106.
- ^ Stevens, Malcolm (1990). Polymer Chemistry An Introduction. Oxford University Press. ISBN 0195057597
- ^ Carothers, Wallace (1936). “Polymers and polyfunctionality”. Transaction of the Faraday Society 32: 39–49. doi:10.1039/TF9363200039.
- ^ Synthetic methods in step-growth polymers. Wiley-Interscience
- ^ Walton, David; Phillip, Lorimer (2000). Polymers. Oxford Univ Pr on Demand. ISBN 0-19-850389-X








