Alkeisreaktio on kemiallinen reaktio, joka tapahtuu yhdessä vaiheessa muodostaen vain yhden siirtymätilan. Reaktion lähtöaineina olevien erilaisten molekyylien lukumäärä viittaa suoraan reaktion molekulaarisuuteen : jos niitä on 1, 2 tai 3 kpl, on tämä alkeisreaktio unimolekulaarinen, bimolekulaarinen tai termolekulaarinen. Molekulaarisuus, joka on pieni positiivinen kokonaisluku, pätee ainoastaan alkeisreaktioille. Alkeisreaktion nopeusvakion dimensiot ovat . Muiden kemiallisten reaktioiden molekulaarisuus ja kertaluku ovat eri asioita.[1][2] Alkeisreaktion differentiaaliyhtälö on yleensä ratkaistavissa ja tämä integroitu yhtälö on reaktion kinetiikan matemaattinen malli. Mallin tarkkuus pitää erikseen todistaa kokeellisilla mittausmenetelmillä tutkimalla reaktion lähtöaineiden kertalukuja.
Sisällys
10. kertaluvun reaktio
2Unimolekulaarinen reaktio
2.1Puoliintumisaika ja keskimääräinen elinaika
3Bimolekulaarinen reaktio, jossa lähtöaineet ovat samanlaiset
4Bimolekulaarinen reaktio, jossa lähtöaineet ovat erilaiset
10Palautuva 1. kertaluvun reaktio etenee tuotteeseen
11Palautuva 2. kertaluvun reaktio
11.1Palautuva 2. kertaluvun reaktio, reaktion alussa vain lähtöaineita
12Huomautukset
13Lisätietoa nimistöstä
14Katso myös
15Lähteet
0. kertaluvun reaktio
Esimerkkinä tästä on suljetussa tilassa tapahtuva reaktio kyllästetyllä pinnalla kuten nesteen pinta tai kiinteän katalyytin pinta. Suljetussa tilassa voi olla nestefaasi, jolta molekyylit desorboituvat kaasufaasiin ja niiden adsorboituminen takaisin nestefaasiin voi tapahtua hyvin hitaasti. Tämänlainen reaktio on heterogeeninen. Reaktiota voidaan tarkastella palautumattomana (irreversiibeli) ja sille voidaan kirjoittaa reaktioyhtälöksi:
Reaktion nopeuslaki on (tässä )
Tässä on reaktionopeus, on stoikiometrinen kerroin, on aineen konsentraatio ja on nopeusvakio. Reaktionopeus ei riipu lähtöaine :n konsentraatiosta. Nopeuslaki on lavennuksen jälkeen kirjoitettavissa muotoon:
, joka on integroitavissa raja-arvoilla ja . Oletetaan, että :n konsentraatio kun on ja kun on .
, jonka ratkaisuksi saadaan
Nopeuslaiksi 0. kertaluvun reaktiolle saadaan:
Kun piirretään xy-koordinaatistoon kuvaaja, jossa x-akselina on aika ja y-akselina on , niin saadaan suora, jonka kulmakerroin on ja leikkauspiste sen y-akselilla on . Kulmakertoimen yksikkö on mol dm-3 s-1SI-yksiköissä.
Unimolekulaarinen reaktio
Tämän reaktio on 1. kertalukua. Esimerkkinä tästä on 137Cs-isotoopin radioaktiivinen hajoaminen. Kaikkien fissioreaktioiden kertaluku on 1. Tämän kertaluvun reaktion reaktiokinetiikka ei riipu lähtöaineen konsentraatiosta. Reaktion nopeuslaki on (tässä ):
1. kertaluvun kinetiikka ja puoliintumisajat.
Sen diffrentiaaliyhtälö voidaan ratkaista integroimalla raja-arvoilla.
, jonka ratkaisu on
, joka on suoran yhtälö, jossa kulmakerroin ja leikkauspiste y-akselilla . Nopeusvakion yksikkö on s-1, eikä 1. kertaluvun kinetiikka ole riippuvainen lähtöaineen konsentraatiosta.
Luonnonlogaritminen yhtälö voidaan muuttaa muotoon
, tai muotoon
, joka kuvaa :n konsentraation eksponentiaalista vähenemistä reaktion edetessä.
Puoliintumisaika ja keskimääräinen elinaika
Integroimalla saadusta luonnonlogaritmisesta yhtälöstä voidaan ratkaista 1. kertaluvun reaktion puoliintumisaika, , merkitsemällä :
Integroimalla saadun eksponenttimuotoisen yhtälön voi kirjoittaa muotoon
Tässä on keskimääräinen elinaika 1. kertaluvun (lähtöaineen konsentraation) vaimenemiselle.
Tässä on :n arvo äärettömän ajan kuluttua. Edellä reaktion nopeusyhtälöstä ja integroimalla ratkaistusta eksponenttimuotoisesta yhtälöstä voidaan kirjoittaa:
Sijoittamalla tämä edelliseen yhtälöön saadaan keskimääräiselle elinajalle integroimalla:
Täten on reaktioaika, jonka kuluessa :n konsentraatio muuttuu .
Bimolekulaarinen reaktio, jossa lähtöaineet ovat samanlaiset
Tämä on 2. kertaluvun reaktio kahden radikaalin (tai ionin,...) kesken. Esimerkkinä on kahden metyyliradikaalin välinen reaktio. Reaktioyhtälö on ():
, yleisesti merkitään
eli , reaktion nopeuslaki on
Integroitu nopeuslaki on
, jonka ratkaisuksi saadaan:
Piirrettäessä xy-koordinaatistoon kuvaaja, jossa y-akselina on ja x-akselina aika , saadaan kulmakertoimeksi ja leikkauspisteestä y-akselilla . Nopeusvakion yksikkö on dm3 mol-1 s-1 ja puoliintumisaika
Bimolekulaarinen reaktio, jossa lähtöaineet ovat erilaiset
Tämä on 2. kertaluvun reaktio kahden erilaisen radikaalin (tai molekyylin,...) kesken. Esimerkkinä on etyyliasetaatin hydrolyysireaktio.
Reaktioyhtälö on:
Reaktion nopeuslaki on ( ja ):
Nopeuslain ratkaisemiseksi voidaan määritellä muuttuja , joka vastaa reaktion etenemistä tuotteeksi:
, nopeuslaki on uudelleenkirjoitettuna:
ja sen integroitu muoto on:
Yhtälön vasen puoli on ratkaistavissa osamurtomenetelmällä ja saadaan
Piirrettäessä yhtälön vasen puoli :n funktiona, niin suoran yhtälöstä kulmakerroin on ja leikkauspiste on . Tästä yhtälöparista voi ratkaista kulmakertoimen arvon.
Pelkistetty reaktiokinetiikka pseudo-olosuhteissa
Joidenkin hydrolyysireaktioiden kinetiikka ei näennäisesti riipu veden määrästä, koska sitä on ylimäärin läsnä. Tämä on esimerkki bimolekulaarisesta reaktiosta pseudo-olosuhteissa, joissa toisen lähtöaineen ylimäärän voi sisällyttää reaktion nopeuslain nopeusvakioon vakiona. Tällöin 2. kertaluvun kinetiikka on yksinkertaistunut 1. kertaluvun pseudo-reaktion kinetiikaksi. Esimerkiksi reaktion
+
nopeuslaki on
Jos :oon verrattuna :ta on ylimäärin, niin sen määrä reaktion kuluessa ei juuri muutu. Tällöin muuttuu :n verran. Nopeuslaille voidaan kirjoittaa:
Koska on lähes vakio, niin nopeuslaki yksinkertaistuu ja noudattaa 1. kertaluvun kinetiikkaa tällä :n konsentraatiolla.
, jossa
Nopeusvakion laskemiseksi, on tehtävä mittaussarja, jossa määritetään :n arvo :n funktiona. Kuvaajan kulmakertoimesta lasketaan reaktion nopeusvakio .
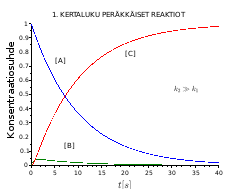
Peräkkäiset 1. kertaluvun reaktiot
Monissa kemiallisissa reaktioissa reaktiotuote lyhytikäinen ja voi reagoida vielä pysyväksi tuotteeksi. Tämänmöinen reaktio etenee välituotteen (intermediaatti) kautta. Kyseessä on kaksi peräkkäin tapahtuvaa 1. kertaluvun alkeisreaktiota.
, jossa B on välituote.
Reaktionopeus A:n vähenemiselle, C:n muodostumiselle ja B:n muutokselle on:
Konsentraatiosuhde on [A]/[A]0 tai [B]/[A]0 tai [C]/[A]0
Reaktion stoikiometriasta saadaa:
Aiemmin osoitettiin, että
Tämän avulla voidaan integroida välituotteen käyttäytymiselle lauseke
Lopputuotteelle C saadaan lauseke käyttämällä A:n ja B:n konsentraatiolausekkeita:
Tarkastelleen vielä välituotetta. Reaktioaika, jolloin välituote B:n konsentraatio on suurimmillaan saadaan derivoimalla sen konsentraatiolauseke ajan suhteen:
On mahdollista vielä säätää reaktio-olosuhteet, missä kosentraation muutos ajan suhteen on hyvin pieni:
(ks. kuva)Konsentraatiosuhde on [A]/[A]0 tai [B]/[A]0 tai [C]/[A]0
Tällöin . Tämä mahdollistaa laskea näissä olosuhteissa välituotteelle ja lopputuotteelle yksinkertaiset integroidut nopeuslait:
Tähän reaktiotilanteeseen on sovellettu vakiotilaoletusta, -oletus, ja se pätee kun . Siis välituote, , ei ehdi merkittävästi kertyä reaktioiden aikana. Tätä -oletusta käytetään usein tilanteissa, missä yksiselitteistä nopeuslakia ei voida ratkaista.
Rinnakkaiset reaktiot
Kemiallisessa kinetiikassa on reaktioita, joissa lähtöaine voi hajota eri tuotteisiin. Tällöin reaktiomekanismissa on useita reaktiokanavia. Esimerkkinä on lähtöaineen hajoaminen 1. kertaluvun nopeuslain mukaisesti tuottaen kolmea eri tuotetta.
ja
ja
Reaktion alussa alkukonsentraatiot ovat . Lähtöaineen hajoamiselle saadaan nopeuslaiksi
Tässä . Yleisesti , on reaktiokanavien lukumäärä. Integroimalla yhtälö saadaan lähtöaineen konsentraation muuttumiselle ajan funktiona
Tuotteelle nopeuslaki on
Integroimalla ja sijoittamalla integrointirajat nopeuslaista saadaan esimerkin kolmelle tuotteelle samalla laskutavalla:
k2 = 3k3= 6k4
Näistä yhtälöistä voidaan päätellä, että nopeusvakioiden suhde saadaan mittaamalla lopputuotteiden konsentraatioiden suhde. Tämä haarautumissuhde ei riipu reaktioajasta:
Tästä haarautumissuhteesta on todettavissa, että se riippuu nopeusvakioista. Sama asia voidaan todeta todennäköisyydellä: mitä suurempi on nopeusvakion arvo sitä todennäköisemmin nopeusvakiota vastaavan reaktiokanavan lopputuote muodostuu reaktiossa. Reaktiokanavan tuotteen saanto, , on määritelty todennäköisyytenä:
Tässä on rinnakkaisreaktion kaikkien reaktiokanavien nopeusvakiot. Kokonaissaannolle pätee , joten saanto :n muodostumiselle .
Jos yhden reaktiokanavan nopeusvakion arvo on moninkertainen verrattuna muihin nopeusvakioihin toisilla reaktiokanavilla, voi näiden tuotteiden käytännön havainnointi vaikeutua johtuen tuotesignaalien pienestä amplitudista.
Kaksi 1. kertaluvun reaktiota samaan tuotteeseen
Tässä tarkastellaan kahta unimolekulaarista reaktiota, joiden lopputuote on sama.
ja
Reaktionopeus lähtöaineiden A1 ja A3 häviämiselle ja lopputuotteen A2 muodostumiselle on
Reaktioiden alussa
Aiemmin osoitettiin, että ja
Lopputuotteen differentiaaliyhtälöksi saadaan
ja integroiduksi nopeuslaiksi saadaan
Palautuva 1. kertaluvun reaktio
Jos reaktion tasapainovakiolle ≈ 1, niin palautuvan reaktion kinetiikka pitää ottaa huomioon nopeuslaissa. Esimerkkinä tällaisesta reaktiosta on cis-1,2-dikloorieteenin ja trans-1,2-dikloorieteenin välinen reaktio. Yksinkertainen tasapainoreaktio on:
Konsentraatiosuhde on [A]/[A]0 tai [B]/[A]0
Reaktion nopeuslaeiksi saadaan:
ja
Reaktion alussa ja
Differentiaaliyhtälöt voidaan ratkaista Laplace muunnoksen ja Cramerin säännön avulla. Ratkaisuksi saadaan
Näistä nopeuslaeista voidaan todeta, että :n eksponentiaalisten vähenemisten nopeusvakio ja :n eksponentiaalisen kasvun nopeusvakio on verrannollinen arvoon . Jos palautuvan reaktion nopeusvakio () on hyvin pieni, niin vaimenee nollaan. Mutta jos on paljon > 0, niin sekä että saavuttavat tasapainokonsentraatiot riittävän pitkän reaktioajan kuluttua. Tällöin tasapainotilassa, , pätee:
Kun tasapainotila on saavutettu, niin reaktionsuuntien reaktionnopeudet ovat yhtäsuuret, joten
Yksittäistasapainoperiaatteen mukaan kun kokonaisreaktio on tasapainotilassa, niin yksittäinen reaktiovaihe on myös tasapainotilassa ja jokaisessa reaktiovaiheessa etenevän reaktiosuunnan kinetiikka on yhtäsuuri kuin tämän reaktiovaiheen palautuvan reaktiosuunnan kinetiikka.
Relaksointimenetelmä
Tasapainotilassa olevan palautuvan (reversiibeli) reaktion kinetiikka voidaan määrittää myös ns. relaksointimenetelmällä.[4] Kun reaktion tasapainotilaa häiritään esim. nopealla lämpötilan nostolla, etenevän ja palautuvan reaktion nopeusvakiot muuttuvat Arrheniuksen yhtälön mukaisesti. Häirinnän seurauksena systeemi palautuu (relaksoituu) uuteen tasapainotilaan ja relaksoitumisaika on mitta palautumiselle. Tarkastellaan relaksointimenetelmää 1. kertaluvun palautuvalla reaktiolla:[5]
Merkitään kuvaamaan lämpötilan noston aiheuttamaa muutosta reaktion ainesosien konsentraatioissa. Tällöin nopeuslaki voidaan kirjoittaa
Integroimalla tämä saadaan:
Merkitään tasapainotilassa ja , joten
Ratkaisemalla tästä ja sijoittamalla ed. logaritmilausekkeeseen saadaan:
Piirrettäessä kuvaaja, jossa y-akselina on ja x-akselina aika , on kulmakertoimen arvo . Tämän käänteisluku on keskimääräinen elinaika, ns. relaksaatioaika:
Relaksaatioaika kuvaa :n eksponentiaalista vaimenemista tai :n eksponentiaalista kasvua uuden tasapainotilan määräämään arvoon. Tasapainotilojen aiheuttaman konsentraatioiden muutos vaimenee :een osaan relaksaatioajan kuluessa. Lisäksi jos tunnetaan tasapainovakion, , suuruus, on nopeusvakiot ratkaistavissa.
Palautuvan reaktion tasapainovakio
Kuten edelle on luettavissa reversiibelin reaktion tasapainovakio, , on reaktiosuuntien nopeusvakioiden suhde, ja myös tuotteiden ja lähtöaineiden konsentraatioiden suhde. Konsentraatioiden osamäärä on myös reaktion ainesosien esiintymistodennäköisyyksien suhde. Olettaen reaktion ainesosat ideaalikaasuiksi tai laimean liuoksen reagensseiksi, tasapainovakiolle voidaan kirjoittaa:
Tässä on tarkasteltavana olevan molekyylin molekulaarinen kokonaisjakaumafunktio.[b] Yhtälössä sekä :lla että :llä on sama vertailukohta kun verrataan niiden energioita. Käytännössä on tätä parempi valita vertailukohdaksi alin energiataso kummallekin molekyylille erikseen. Tällöin on energiaero :n alimman energiatason ja :n alimman energiatason välillä. Nyt Boltzmannin lain[6] mukaan tasapainovakiolle voidaan kirjoittaa:[c]
Tässä on Boltzmannin vakio ja lämpötila Kelvin-asteissa.
Jos reaktion ainesosien kokonaisjakaumafunktiot voidaan laskea, niin reaktion tasapainovakio on laskettavissa.
Palautuva 1. kertaluvun reaktio etenee tuotteeseen
Vakiotilaoletuksen käyttö kompleksisen reaktion kinetiikan ratkaisemisessa korostuu 1. kertaluvun reaktiossa, jossa on mukana välituote. Reaktiomekanismi jota tarkastellaan on[7]
Reaktion nopeuslait ovat:
Konsentraatiosuhde on [A]/[A]0 tai [B]/[A]0 tai [C]/[A]0
Yleinen ratkaisu esim. Laplace muunnoksen avulla näille differentiaaliyhtälöille on
Näissä yhtälöissä . Viereisessä kuvassa on tilanne, jossa ja . Lopputuotteen muodostuminen viivästyy johtuen lähtöaineen ja välituotteen palautuvasta (reversiibelistä) reaktiosta. Jos olisi erittäin pieni, niin reaktiot olisivat kuten peräkkäiset 1. kertaluvun reaktiot.
Sovellettaessa vakiotilaoletusta reaktio välituotteelle, saadaan sen konsentraatioksi
Tärkeätä on huomata, että välituotteen konsentraation pitää olla paljon pienempi kuin lähtöaineen konsentraation, joten . Vakiotilaoletusta soveltaen saadaan edellä mainituista tarkkoista integroiduista nopeuslaeista approksimoidut yhtälöt:
Viereisestä kuvasta on todettavissa, että approksimoidut yhtälöt lähestyvät tarkkoja yhtälöitä vasta riittävän pitkän reaktioajan kuluttua: .
Konsentraatiosuhde on [A]/[A]0 tai [B]/[A]0 tai [C]/[A]0
Tätä aikaa voi lyhentää ottamalla nopeusvakio hyvin pieneksi kuten . Vakiotilaoletuksen edellys on, että välituotteen konsentraatio on aina hyvin pieni tai se muuttuu reaktioajan funktiona hyvin hitaasti. Riippuen nopeusvakioiden suuruudesta, joko ensimmäinen reaktiovaihe (palautuva) tai toinen reaktiovaihe (palautumaton) voi olla kokonaisreaktionopeuden määräävä vaihe. Jos , niin tuotteen muodostumiselle voidaan kirjoittaa
Toisaalta jos on hyvin suuri, jolloin toinen reaktiovaihe on liian hidas vaikuttaakseen merkittävästi tasapainotilaan , niin tuotteen muodostumiselle saadaan
Tällöin toinen reaktiovaihe on kokonaisreaktionopeuden määräävä vaihe.
Palautuva 2. kertaluvun reaktio
Jos bimolekulaarisen reaktion palautuva reaktiosuunta on kinetiikaltaan merkittävä, pitää se ottaa huomioon nopeuslaissa. Tällöin on kysy palautuvasta 2. kertaluvun reaktiosta. Tasapainoreaktio on:
Reaktioyhtälön nopeuslaki on
Tässä on termodynaaminen tasapainovakio. Merkitään reaktion etenemistä :llä, jolloin nopeuslaiksi saadaan
Palautuva 2. kertaluvun reaktio, reaktion alussa vain lähtöaineita
Jos reaktion alussa vain ja ovat läsnä, niin nopeuslaki on
Merkitään nyt . Nyt nopeuslaki on kirjoitettavissa muotoon
ja se on em. tavoin integroitavissa. Kun lisäksi reaktion alussa ja , niin ratkaisuksi saadaan edellä oleva yhtälö. Se voidaan kirjoittaa myös muotoon:
Tässä yhtälössä .
Jos lähtöaineiden alkukonsentraatiot ja tasapainovakio tunnetaan, voidaan laskea. Nopeusvakio saadaan kuvaajasta jossa yhtälön vasen puoli on piirretty ajan funktiona.
Huomautukset
↑Vaihtoehtoisesti keskimääräinen elinaika saadaan jakaumafunktiosta . Tässä on normitusvakio, joka takaa kokonaistodennäköisyydeksi arvon 1.[3]
Tästä saadaan . Keskimääräisen elinajan odotusarvo on määritelmää mukaellen
↑Molekulaarinen kokonaisjakaumafunktio koostuu translaatio- (liike), rotaatio- (pyöriminen), vibraatio- (värähtely), ja elektronijakaumafunktion tulosta.
↑Tarkasteltaessa hyvin suurta joukkoa hiukkasia, on Boltzmannin jakaumalain mukaan todennäköisin jakauma mille tahansa energiatilajoukolle, joille voidaan sijoittaa -kappaletta näitä hiukkasia kokonaisenergialla , suurin mahdollinen permutaatioiden lukumäärä tässä jakaumassa. Tälle todennäköisimmälle jakaumalle, jossa on -kappaletta hiukkasia energialla lämpötilassa , jakaumalain mukaan voidaan kirjoittaa
Energialtaan yhtä suuret energiatilat huomioidaan tilastollisella painolla (degeneraatiolla) :
Tässä on molekulaarinen jakaumafunktio. Tarkasteltaessa yhtä molekyyliä suuresta joukosta molekyylejä termisessä tasapainotilassa on se tietyllä energiatasolla energialla ja tilastollisella painolla todennäköisyydellä, joka on verrannollinen arvoon , jossa on absoluuttinen lämpötila ja on Boltzmannin vakio. Todennäköisyys sille, että se esiintyy millä tahansa sen mahdollisista energiatiloista on verrannollinen arvoon
Lisätietoa nimistöstä
Reaktion kinetiikkaan liittyviä käsitteitä IUPAC:n mukaan





![{\displaystyle R=-{\frac {1}{a}}{\frac {d[A]}{dt}}=k[A]^{0}=k}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1d54cdc21772afa96e14b41ff7fa1afbfe31b10f)

![{\displaystyle [A]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/79eaa334597b1861f1b08ca0c8fecb3858ebcb12)


![{\displaystyle d[A]=-k\,dt}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e269d25b9a5b3aedcbe1b0bcf93920bb55121da0)



![{\displaystyle [A]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3213f44359fda514400d0682cec94cdc05ac1f4c)

![{\displaystyle [A]_{t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/03d6eaf7fc064571a2d2e88332f923d25a436f1c)
![{\displaystyle \int _{[A]_{0}}^{[A]_{t}}d[A]=-k\int _{t_{1}=0}^{t_{2}=t}dt}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6992a123fb77e63734a2ab0a7c81dde6b9d27b90)
![{\displaystyle [A]_{t}-[A]_{0}=-k(t-0)}](https://wikimedia.org/api/rest_v1/media/math/render/svg/47e58573e08069db9bc3cfc379a78334b1af770a)
![{\displaystyle [A]_{t}=[A]_{0}-kt}](https://wikimedia.org/api/rest_v1/media/math/render/svg/feb953bcb1af029c02c466cbde643e92a810adea)



![{\displaystyle R=-{\frac {1}{a}}{\frac {d[A]}{dt}}=k[A]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/64328266eff53dae1cf48230fe89ad122a772d7a)
![{\displaystyle \int _{[A]_{0}}^{[A]_{t}}{\frac {d[A]}{[A]}}=-a\,k\int _{0}^{t}dt}](https://wikimedia.org/api/rest_v1/media/math/render/svg/baa9c8f2745016cf60d27821e1149e1bf5870e56)
![{\displaystyle \ln[A]_{t}=\ln[A]_{0}-a\,k\,t}](https://wikimedia.org/api/rest_v1/media/math/render/svg/31ee42524c7a9b8213d8588b8e92e805f0271ed6)

![{\displaystyle \ln[A]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/94c3eceff30335654c49c85848e9a147d4260c21)
![{\displaystyle \ln {\Bigg (}{\frac {[A]_{t}}{[A]_{0}}}{\Bigg )}=-a\,k\,t}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8897bdab479e7b29402aeff2f10bbff9c5337f84)
![{\displaystyle [A]_{t}=[A]_{0}e^{-a\,k\,t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9d3ab1720d40a5e3ccae21f94e72b78449bfe702)

![{\displaystyle [A]_{t}={\frac {[A]_{0}}{2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d9c73e7505113af9161a4dc7226f89479ee2fdbf)
![{\displaystyle \ln {\frac {[A]_{0}}{2}}=-a\,k\,t_{1/2}+\ln[A]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/13aa7b5cf791a6706b767a057b57d969c5050286)
![{\displaystyle t_{1/2}\,=\,{\frac {\ln {\frac {[A]_{0}}{2[A]_{0}}}}{-a\,k}}\,=\,{\frac {\ln 2}{a\,k}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/eb8a7a7093c7a81c2c8a21a3f7915cfaf98df461)
![{\displaystyle [A]_{t}\,=\,[A]_{0}e^{-a\,k\,t}\,=\,[A]_{0}e^{-{\frac {t}{\tau }}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0bfa08cf90e7cc9e60e34c77c483c40c6313f8b7)

![{\displaystyle \left\langle t\right\rangle \,=\,{\frac {\int _{[A]_{0}}^{[A]_{\infty }}t\,d[A]}{\int _{[A]_{0}}^{[A]_{\infty }}d[A]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4eb89ec774dcfd257b5916ea7d283b2f186cba43)
![{\displaystyle [A]_{\infty }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b5353029bc582be367c568abaf932f3961101059)
![{\displaystyle d[A]\,=\,-a\,k[A]_{0}e^{-a\,k\,t}dt}](https://wikimedia.org/api/rest_v1/media/math/render/svg/820d0d757f2eabbc3b4f518f4610a14a52d2175a)

![{\displaystyle [A]_{0}\to {\frac {[A]_{0}}{e}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/96823fa18f282151ebc7b6218287ab159d774e7c)




![{\displaystyle R=-{\frac {1}{a}}{\frac {d[A]}{dt}}=k[A]^{2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ba3a6d082979e0cb40182b0c5e94511a221dcda7)
![{\displaystyle \int _{[A]_{0}}^{[A]_{t}}{\Bigg (}-{\frac {d[A]}{[A]^{2}}}{\Bigg )}=2k\int _{0}^{t}dt}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8ddcf2b2723136334221c28ed46c958afbd40db7)
![{\displaystyle {\frac {1}{[A]_{t}}}-{\frac {1}{[A]_{0}}}=2k\,t}](https://wikimedia.org/api/rest_v1/media/math/render/svg/77133b3ca4bc169d7348f465a3bd3a57443fd1c7)
![{\displaystyle {\frac {1}{[A]_{t}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5c670432f85b8baadaaa855d38f70bac82e25c8b)

![{\displaystyle {\frac {1}{[A]_{0}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2e5be2bede9054fb5b5ee04bb9f9d30f81543c2a)
![{\displaystyle t_{1/2}={\frac {1}{2k[A]_{0}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c2009de2148b52bdedaafdb5dc3f6b897ae8c1d5)




![{\displaystyle R=-{\frac {d[A]}{dt}}=k[A]^{1}[B]^{1}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/021d0d7a6f8dc836eac502a78bd02291df9fd036)

![{\displaystyle x={\Big (}[A]_{0}-[A]_{t}{\Big )}={\Big (}[B]_{0}-[B]_{t}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/07154164a2d501682a48da9546c32f35a2f492d1)
![{\displaystyle {\frac {dx}{dt}}=k{\Big (}[A]_{0}-x{\Big )}{\Big (}[B]_{0}-x{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/54381af5ca8c7798ca4ec65f47d3b61efd28afab)
![{\displaystyle \int _{x(0)}^{x(t)}{\frac {dx}{{\Big (}[A]_{0}-x{\Big )}{\Big (}[B]_{0}-x{\Big )}}}=k\int _{0}^{t}dt}](https://wikimedia.org/api/rest_v1/media/math/render/svg/10fe7ed7292678607925b969a30df62debc61545)
![{\displaystyle \ln {\frac {{\Big (}[A]_{0}-x{\Big )}}{{\Big (}[B]_{0}-x{\Big )}}}=k{\Big (}[A]_{0}-[B]_{0}{\Big )}t+\ln {\frac {[A]_{0}}{[B]_{0}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/073e29d9d33e088be096ed1751f3b32a6edee326)
![{\displaystyle k{\Big (}[A]_{0}-[B]_{0}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/49dbc7c90285dfae3ffc2ec7b43f34fff7502b1f)
![{\displaystyle \ln {\Bigg (}{\frac {[A]_{0}}{[B]_{0}}}{\Bigg )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ad6cb83ca3528ffbf03c468e740ddfaa41ac013c)

![{\displaystyle -{\frac {d[A]}{dt}}=k[A]^{\alpha }[B]^{\beta }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8961c974101f59cb98cba80023904175dc1988e6)
![{\displaystyle [B]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6e6c6caf9d188e4a608dfdac98bbcd140a9ea106)
![{\displaystyle -{\frac {d[A]}{dt}}={\frac {dx}{dt}}=k([A]_{0}-x)^{\alpha }[B]^{\beta }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a607cbc050391c15d6b1ac86b232e92afc8993ef)

![{\displaystyle {\frac {dx}{dt}}=k'([A]_{0}-x)^{\alpha }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e8822e919fb840e2911416e163f360c61e2da379)
![{\displaystyle k'=k[B]^{\beta }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/996b55c8f541d7985cde8c93bb8a1c0709db82f5)

![{\displaystyle {\ce {A {\ce {->[k_1]}}B {\ce {->[k_2]}}C}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/45fa2ef3df65cded96533ff05c0fb621714e5ad3)

![{\displaystyle R=-{\frac {d[A]}{dt}}=k_{1}[A]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1f487b90cfe43d77e6aad687a4083871e9000277)
![{\displaystyle {\frac {d[B]}{dt}}=k_{1}[A]-k_{1}[B]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/061c7b0f4bd7ece4765d2106e349c08d6038fe94)
![{\displaystyle {\frac {d[C]}{dt}}=k_{2}[B]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/fe4910c6054aef88e763916ed2fd70380eb01eb3)
![{\displaystyle [A]+[B]+[C]=[A]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0a7bcb70deda2b099aafbd5650e06ce1e37e206b)
![{\displaystyle [A]=[A]_{0}e^{-k\,t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d59c47f6889146f01410b40ff0310e10d4f10a33)

![{\displaystyle [B]={\frac {k_{1}[A]_{0}}{k_{2}-k_{1}}}{\Big (}e^{-k_{1}t}-e^{-k_{2}t}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/561ad821167d503bc9822936178c08e9411b9708)
![{\displaystyle [C]=[A]_{0}{\Bigg (}1-{\frac {1}{k_{2}-k_{1}}}{\Big (}k_{2}e^{-k_{1}t}-k_{1}e^{-k_{2}t}{\Big )}{\Bigg )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9cb646115f9f848a0cd9a4ed014d566a2959b23a)
![{\displaystyle {\Bigg (}{\frac {d[B]}{dt}}{\Bigg )}_{t=t_{max}}=0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/582435a4c2a9527a5f27b53ce31a2c7da9814bca)

![{\displaystyle {\frac {d[B]}{dt}}=-k_{1}[B]+k_{1}[A]=0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5d6e14dba6ba28d5fc979d56fba586f00f983792)

![{\displaystyle [B]_{_{\text{VT}}}={\frac {k_{1}}{k_{2}}}[A]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f0d50ddb384ae23231ef28e76c1487a1d3109177)
![{\displaystyle [B]_{_{\text{VT}}}={\frac {k_{1}}{k_{2}}}[A]_{0}\,e^{-k_{1}\,t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/785b27b76785e00955d794daf780c11fe56296f8)
![{\displaystyle [C]_{_{\text{VT}}}=[A]_{0}\,{\Big (}1-e^{-k_{1}\,t}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/195089bfa8d1f8c39386d90fb4d291c227b40967)



![{\displaystyle {\ce {A_1 {\ce {->[k_2]}}A_2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/933e6a44c9362b81fd77d7c4f3aad1439196e1b0)
![{\displaystyle {\ce {A_1 {\ce {->[k_3]}}A_3}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/158968c94d13e913ebe84cddbda49026bc93009b)
![{\displaystyle {\ce {A_1 {\ce {->[k_4]}}A_4}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f77c3a28fba035c8d50ceed625248890468f606d)
![{\displaystyle [A_{1}]=[A_{1}]_{0},[A_{2}]=[A_{3}]=[A_{4}]=0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d15d5b8b4bdfb8b8a189ecefe9b73f12408f7460)
![{\displaystyle R\,=\,-{\frac {d[A_{1}]}{dt}}=k_{2}[A_{1}]+k_{3}[A_{1}]+k_{4}[A_{1}]=k_{s}[A_{1}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f965ec9b5f17980d75ff99643f1ecb5ceade4ac8)



![{\displaystyle [A_{1}]\,=\,[A_{1}]_{0}\,e^{-k_{s}\,t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/04674e1d5fb62cbd78922133bb418a2f4eec6c30)
![{\displaystyle R=+{\frac {d[A_{2}]}{dt}}\,=\,k_{2}[A_{1}]\,=\,k_{2}[A_{1}]_{0}\,e^{-k_{s}t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ec94623cab95e86292e5bd001986c96ada93c578)

![{\displaystyle [A_{2}]\,=\,{\frac {k_{2}[A_{1}]_{0}}{k_{s}}}{\Big (}1-e^{-k_{s}t}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0758f925e9518ea3c84680c7c2fd356b1eb025ea)
![{\displaystyle [A_{3}]\,=\,{\frac {k_{3}[A_{1}]_{0}}{k_{s}}}{\Big (}1-e^{-k_{s}t}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b1ae430e55227fdccbc770e219fc0cd6cd6403a5)
![{\displaystyle [A_{4}]\,=\,{\frac {k_{4}[A_{1}]_{0}}{k_{s}}}{\Big (}1-e^{-k_{s}t}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/357c2517b1138ab1412f9a03cf1bf7049ae768ed)
![{\displaystyle {\frac {[A_{3}]}{[A_{4}]}}\,=\,{\frac {k_{3}}{k_{4}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e072793295fb6e8f14cf97c06ed6bd26e55de908)




![{\displaystyle [A_{4}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/43d600a6ad621e63fad888510d6f2e53ce9f060c)
![{\displaystyle \Phi _{[A_{4}]}={\frac {1}{10}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0719ad77b075d738ba9ddd09e72b7db5e8efcb62)
![{\displaystyle {\ce {A_1 {\ce {->[k_1]}}A_2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e772abe2ac5cdb140bff3d2f133df30c14864a0a)
![{\displaystyle {\ce {A_3 {\ce {->[k_3]}}A_2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/49fb926ae3d0ab8669bff881b459610e97cb0b04)
![{\displaystyle -{\frac {d[A_{1}]}{dt}}=k_{1}[A_{1}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3f1008d309a47edb1d6e7454cb9edb70ff162e91)
![{\displaystyle +{\frac {d[A_{2}]}{dt}}=k_{1}[A_{1}]+k_{3}[A_{3}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9877360cc96f5d0e0c89d200f944c741807d2799)
![{\displaystyle -{\frac {d[A_{3}]}{dt}}=k_{3}[A_{3}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7f029837c3017646ab41ce7387cb916039bbedd2)
![{\displaystyle [A_{1}]=[A_{1}]_{0},\,[A_{3}]=[A_{3}]_{0},\,[A_{2}]=0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5b61610f47987d613979181f3c200fe41b5ee074)
![{\displaystyle [A_{1}]=[A_{1}]_{0}e^{-k_{1}\,t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6b2c13e7e8c14355e6c81ecda85d2648e6dbc920)
![{\displaystyle [A_{3}]=[A_{3}]_{0}e^{-k_{3}\,t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/645bd21010c0261c2e8bf420c6e75fc441107921)
![{\displaystyle R=+{\frac {d[A_{2}]}{dt}}=k_{1}[A_{1}]_{0}e^{-k_{1}\,t}+k_{3}[A_{3}]_{0}e^{-k_{3}\,t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5e57d1e9bf15c2aa654b3b6aaa08ad78166dee1d)
![{\displaystyle [A_{2}]=[A_{1}]_{0}-[A_{1}]_{0}e^{-k_{1}\,t}+[A_{3}]_{0}-[A_{3}]_{0}e^{-k_{3}\,t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/366371d18482c3370b06b38f6f2d16a68f64979d)



![{\displaystyle -{\frac {d[A]}{dt}}=k_{1}[A]-k_{-1}[B]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9c1150cf708084d9374deb4aeb780ce1bbcab4fc)
![{\displaystyle -{\frac {d[B]}{dt}}=k_{-1}[B]-k_{1}[A]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e3201c59bcfbded3ede738ed4bd60ce6281e34a2)
![{\displaystyle [A]=[A]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/40eb08020bb643f51becae46371f5e8afd9bca33)
![{\displaystyle [B]=0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/dcff442d7499cc7c6c2cb177bbe5788e6559f915)
![{\displaystyle [A]={\frac {[A]_{0}}{k_{1}+k_{-1}}}{\Big (}k_{-1}+k_{1}e^{-(k_{1}+k_{-1})t}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ccd755280b1ca4cbfa94fd510ab36031dd118f63)
![{\displaystyle [B]={\frac {k_{1}[A]_{0}}{k_{1}+k_{-1}}}{\Big (}1-e^{-(k_{1}+k_{-1})t}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0ec64320790ee5fcaeaffe81e54c97ce6e12ff25)



![{\displaystyle [A]_{\text{tp}}=\lim _{t\to \infty }[A]=[A]_{0}{\frac {k_{-1}}{k_{1}+k_{-1}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8168e2e32bc794cb6d96c179172bcdba4b36d33e)
![{\displaystyle [B]_{\text{tp}}=\lim _{t\to \infty }[B]=[A]_{0}{\Big (}1-{\frac {k_{-1}}{k_{1}+k_{-1}}}{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/aa535275eebf9e28034caf4518e81f3ba522eaf6)
![{\displaystyle {\frac {d[A]_{\text{tp}}}{dt}}={\frac {d[B]_{\text{tp}}}{dt}}=0=-k_{1}[A]_{\text{tp}}+k_{-1}[B]_{\text{tp}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/cf5c2aece7121f3709f4192f4f01b3e82f52efa9)
![{\displaystyle {\frac {k_{1}}{k_{-1}}}={\frac {[B]_{\text{tp}}}{[A]_{\text{tp}}}}=K}](https://wikimedia.org/api/rest_v1/media/math/render/svg/402c96b99de30a730cb93554e9935212ea98c416)
![{\displaystyle {\frac {dx}{dt}}=k_{1}([A]_{0}-x)-k_{-1}([B]_{0}+x)}](https://wikimedia.org/api/rest_v1/media/math/render/svg/29a45e79c96eaca2245c939600d97db482f09f80)
![{\displaystyle \int _{0}^{x}{\frac {dx}{k_{1}[A]_{0}-k_{-1}[B]_{0}-(k_{1}+k_{-1})x}}=\int _{0}^{t}dt}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2f59544221d4d3a7a71cd55cbc9d923f3017afe8)
![{\displaystyle \ln {\Bigg (}{\frac {k_{1}[A]_{0}-k_{-1}[B]_{0}}{k_{1}[A]_{0}-k_{-1}[B]_{0}-(k_{1}+k_{-1})x}}{\Bigg )}=(k_{1}-k_{-1})t}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9490118086882404572d4dc8b4636721a54b8a43)


![{\displaystyle k_{1}([A]_{0}-x_{e})=k_{-1}([B]_{0}+x_{e})}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c9b12fd5c287cff77b5a202f270a3e201770cc86)
![{\displaystyle k_{1}[A]_{0}-k_{-1}[B]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/33fad3f0dfce3604ae620d1768a0350340b45209)





![{\displaystyle K={\frac {[B]_{\text{tp}}}{[A]_{\text{tp}}}}={\frac {Q_{B}}{Q_{A}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/59746f6d55c6469d06aef1a0480bf4743e6385b2)








![{\displaystyle +{\frac {d[A]}{dt}}=-k_{1}[A]+k_{-1}[B]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e6831c0e65d64b92c9a959c6d0bb60c3662e95db)
![{\displaystyle +{\frac {d[B]}{dt}}=k_{1}[A]-(k_{-1}+k_{2})[B]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a8aa3c13e9ba8d78ea5ea5aa67d4e8f758668d2a)
![{\displaystyle +{\frac {d[C]}{dt}}=k_{2}[B]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7f16bec09c98d778c7c4f63e4bafa71f11706809)
![{\displaystyle [A]\,=\,{\frac {[A]_{0}}{\lambda _{2}-\lambda _{3}}}{\bigg (}(k_{1}-\lambda _{3})\,e^{-\lambda _{2}\,t}-(k_{1}-\lambda _{2})e^{-\lambda _{3}\,t}{\bigg )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1157e336e3f49aecf76e4ed57109b4cb461e8840)
![{\displaystyle [B]\,=\,{\frac {[A]_{0}k_{1}}{\lambda _{2}-\lambda _{3}}}{\bigg (}-e^{-\lambda _{2}\,t}+e^{-\lambda _{3}\,t}{\bigg )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9fa331d1490a743a5da90e3f9ff82b66231370b0)
![{\displaystyle [C]\,=\,[A]_{0}{\bigg (}1+{\frac {\lambda _{2}\,\lambda _{3}}{(\lambda _{2}-\lambda _{3})}}\,{\frac {1}{\lambda _{2}}}\,e^{-\lambda _{2}\,t}+{\frac {1}{\lambda _{3}}}\,e^{-\lambda _{3}\,t}{\bigg )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/392af87a74541cf3b2de180bf786d5bb7dd64c6c)



![{\displaystyle [B]_{_{VT}}\,=\,[A]{\frac {k_{1}}{k_{-1}+k_{2}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4038626e8477369465e192ce5343333f3f655412)

![{\displaystyle [A]\,=\,{\frac {[A]_{0}}{k_{-1}+k_{2}}}{\bigg (}{\frac {k_{1}k_{-1}}{k_{-1}+k_{2}}}\,e^{-(k_{-1}+k_{2})\,t}+(k_{-1}+k_{2})\,e^{-{\frac {k_{1}k_{2}}{k_{-1}\,+\,k_{2}}}\,t}{\bigg )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/60f00693905b9d1a0cd4346ddabf47d893515b23)
![{\displaystyle [B]\,=\,{\frac {[A]_{0}\,k_{1}}{k_{-1}+k_{2}}}{\bigg (}-e^{-(k_{-1}+k_{2})\,t}+\,e^{-{\frac {k_{1}k_{2}}{k_{-1}\,+\,k_{2}}}\,t}{\bigg )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6976713fae2322a85c37b5265fcaf183e18f3676)
![{\displaystyle [C]\,=\,[A]_{0}{\Bigg [}1+{\frac {k_{1}k_{2}}{k_{-1}+k_{2}}}{\Bigg (}{\frac {1}{k_{-1}+k_{2}}}\,e^{-(k_{-1}+k_{2})\,t}-{\frac {k_{-1}+k_{2}}{k_{1}k_{2}}}\,e^{-{\frac {k_{1}k_{2}}{k_{-1}+k_{2}}}\,t}{\Bigg )}{\Bigg ]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b68e35eff908687e631449db468dba1efcd4d18b)





![{\displaystyle +{\frac {d[C]}{dt}}\,\simeq \,k_{1}[A]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1e8136e39713c5298f22b458d848fbc87925f9dd)


![{\displaystyle +{\frac {d[C]}{dt}}\,\simeq \,{\frac {k_{1}k_{2}}{k_{-1}}}[A]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3b1d00cdd71ebe440fd96876b10255b0f00068d2)

![{\displaystyle -{\frac {d[A]}{dt}}\,=\,k_{1}[A][B]\,-\,k_{-1}[C][D]\,=\,k_{1}{\Bigg (}[A][B]\,-\,{\frac {1}{K}}C][D]{\Bigg )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/69e30f0f092daf4506bab2dd3be01f9aadf0611b)
![{\displaystyle {\frac {dx}{dt}}\,=\,k_{1}{\Big (}[A]_{0}-x{\Big )}{\Big (}[B]_{0}-x{\Big )}\,-\,k_{-1}{\Big (}[C]_{0}\,+\,x{\Big )}{\Big (}[D]_{0}\,+\,x{\Big )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8d9fee57fd8c96c6f0e61b63ec08d84526269cbc)
![{\displaystyle \alpha =(k_{1}[A]_{0}[B]_{0}-k_{-1}[C]_{0}[D]_{0}),\,\,\,\beta =-[k_{1}([A]_{0}+[B]_{0})+k_{-1}([C]_{0}+[D]_{0})],\,\,\,\gamma =(k_{1}+k_{-1})}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5ee1d14b0acc23cd6957d3d34f6e365a7f7361fe)



![{\displaystyle {\frac {1}{k_{1}}}{\frac {dx}{dt}}\,=\,{\Big (}[A]_{0}-x{\Big )}{\Big (}[B]_{0}-x{\Big )}\,-\,{\frac {x^{2}}{K}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6426af15a6d5cf2b02586c75bdf6e5f598ecfcc4)
![{\displaystyle \alpha =([A]_{0}[B]_{0}),\,\,\,\beta =-([A]_{0}+[B]_{0}),\,\,\,\gamma =(1-K^{-1})}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d8ef94babfcd7cdcab617de9800f9d3782454ec1)



![{\displaystyle {\frac {1}{\sqrt {q}}}\ln {\Bigg [}{\frac {(2\gamma \,x)/(\beta \,-\,{\sqrt {q}})+1}{(2\gamma \,x)/(\beta \,+\,{\sqrt {q}})+1}}{\Bigg ]}\,=\,k_{1}t}](https://wikimedia.org/api/rest_v1/media/math/render/svg/98d9c41bb5fa0db7d57cd2d4d39eb7e1ba313577)


![{\displaystyle P(t)=C[A]_{0}e^{-kt}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e2fbf10b23a37e2d673cba5c4eb0b682fba4566a)

![{\displaystyle 1\,=\,\int _{0}^{\infty }C\,[A]_{0}e^{-kt}dt\,=\,C{\frac {[A]_{0}}{k}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/05c88d1886d23b980321557d1241b42b7c6ed6a4)
![{\displaystyle C={\frac {k}{[A]_{0}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/50bff3a6759241a0b8c28dfad359813308c1d206)
![{\displaystyle \left\langle t\right\rangle \,=\,\int _{0}^{\infty }tP(t)dt\,=\,\int _{0}^{\infty }t\,C[A]_{0}e^{-kt}dt\,=\,{\frac {1}{k}}\,=\,\tau }](https://wikimedia.org/api/rest_v1/media/math/render/svg/d85d400b50d9373c89f0c57a0eae0e46b5339d99)
















